Acromegalia: Diagnóstico y Tratamiento
Carlos Eduardo Jimenez - Canizales
Directo Grupo de Investigación Ibagué Saludable, Ibagué, Tolima
Director Dimetabolismo, Mariquita, Tolima
Especialista en Medicina Interna, Universidad Surcolombiana, Neiva, Huila
Fellow de endocrinología, Fundación Universitaria de Ciencias de la Salud, Bogotá
Introducción
Mediante la presentación de un caso de la experiencia del Hospital de San José y la Fundación Universitaria de Ciencias de la Salud (FUCS)se plantearon las estrategias actuales de diagnostico y manejo de los pacientes con acromegalia y se realizó una revisión a modo de mesa de trabajo con los residentes de endocrinología asistente a la actividad a continuación se relata los componentes teóricos revisados.
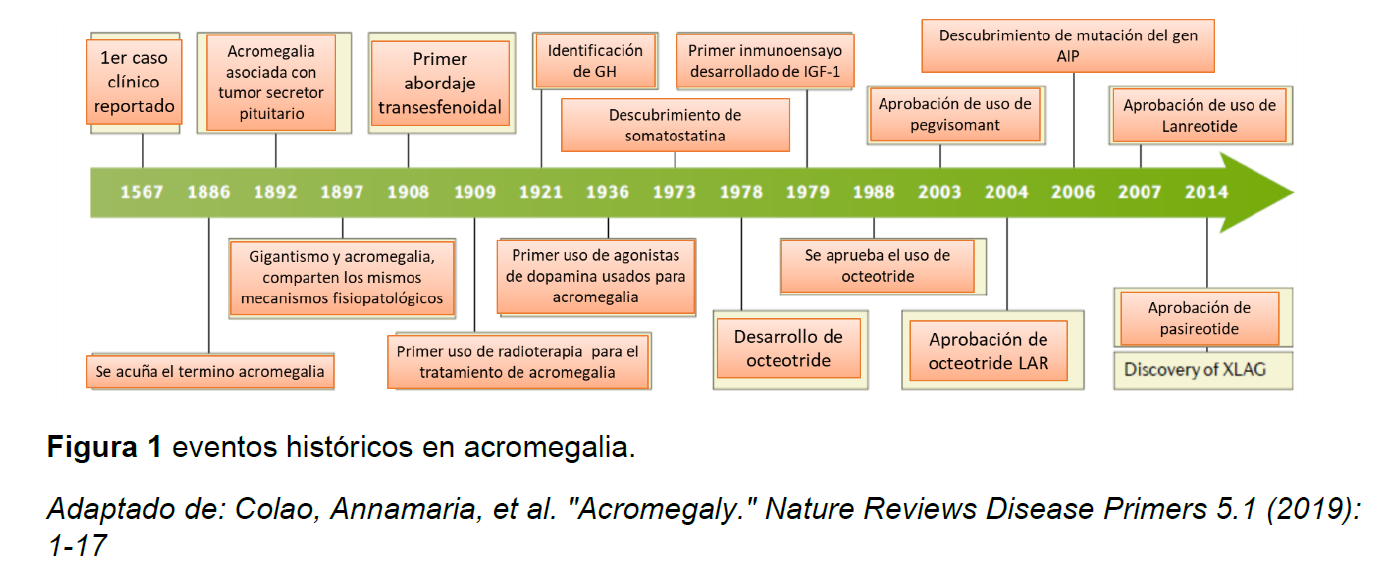
La historia de la acromegalia se remota a eventos históricos reportados en textos egipcios y romanos en donde se considera que personajes importantes de la historia como el faraón Akenathon tenía rasgos de acro-gigantismo y el general romano Maximinus Thrax en los bustos lucia con rasgos de sugestivos de crecimiento acral, el primer caso formal se reporta en 1567 y es en 1886 que se acuña el termino de acromegalia para referirse a una condición clínica de crecimiento excesivo en el adulto, posteriormente se identifican los fenómenos fisiopatológicos involucrados y su igualdad con gigantismo en momentos de crecimiento distintos, la relación con patología hipofisiaria y secuencialmente una serie de eventos científicos que en la actualidad nos permiten de forma selectiva dar manejo no solo quirúrgico si no de bloqueo hormonal con análogos de somatostatina y bloqueante del receptor de GH como el pegvisomant(Ver figura 1) (1-4).
La epidemiologia de la acromegalia es muy diversa dependiendo del perfil de la cohorte evaluada, sin tener una correlación clara con raza, género o etnia, sin embargo, se plantea que la media de edad es de 44 años, con incidencia de alrededor de 3-4 por millón por año y una prevalencia de 60 por millón y con un porcentaje bajo asociado a otras condiciones como hiperprolactinemia (25%), síndrome de carney (PRKAR1A), McCune-Albright (GNAS), Neoplasia endocrina múltiple (NEM) tipo 1 y neoplasias productoras de hormona del crecimiento (HC) o factor de crecimiento similar a la insulina tipo 1 (IGF-1).
Perlas para la práctica clínica
Diferenciación de linajes celulares
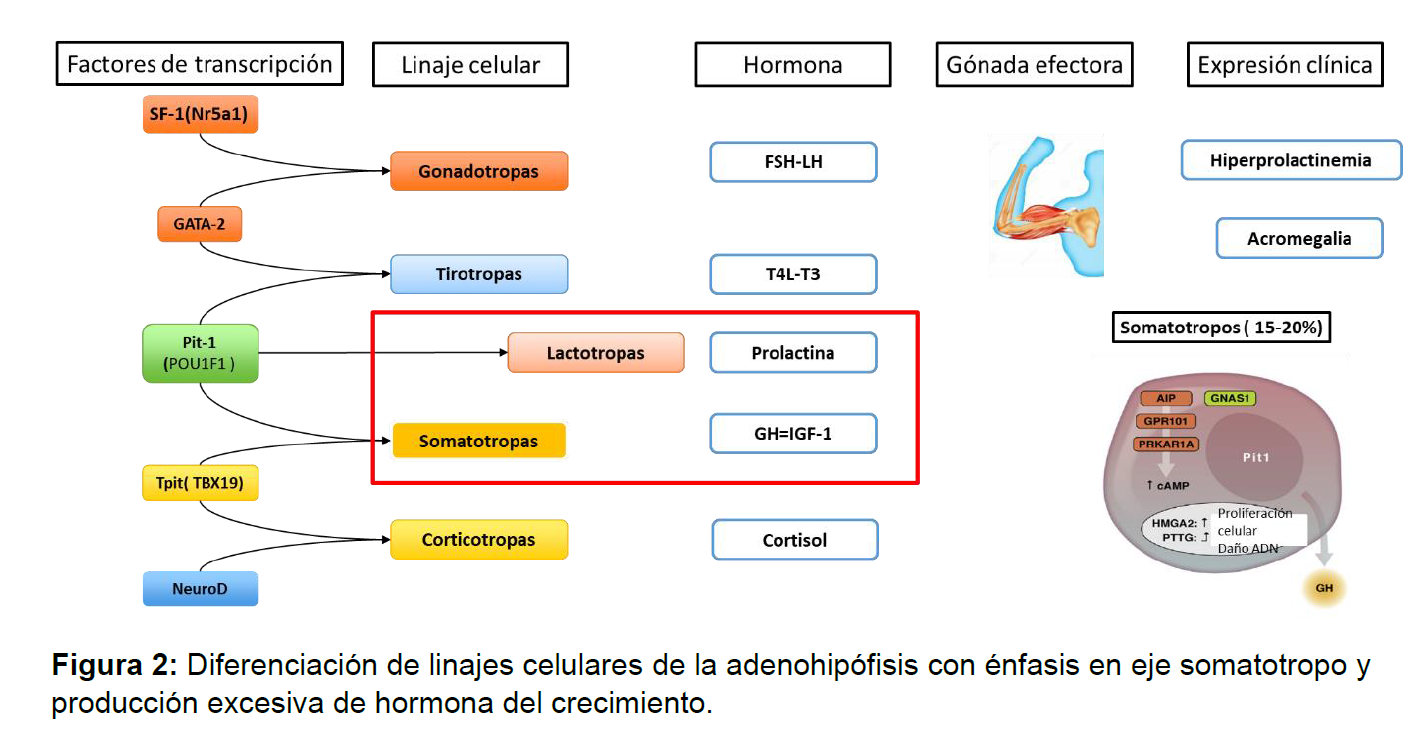
El entendimiento de las embriología e histología de la adenohipófisis a permitido no solo cambias los paradigmas de clasificación de los tumores hipofisiarios a tumores neuroendocrinos hipofisiarios, sino, además permitir una clasificación no solo histológica si no terapéutica y pronostica, aportando al riesgo de tener una mejor respuesta a ciertos fármacos y planteando un mayor riesgo de recaída, por lo cual entender los factores de transcripción implicados y su expresión hormonal es fundamental para el clínico(ver figura 2)(5, 6).
Etiología
Mas del 90 % son tumores neuroendocrinos hipofisiarios (PitNet) antes conocidos principalmente como adenomas hipofisiarios y los cuales se clasificaban exclusivamente con fines quirúrgicos como micro-adenomas (<10 mm) y macro-adenomas(>10mm) clasificación que actualmente esta relejada como imagenológica ya que una vez se defina la funcionalidad hormonal se reclasifican en PitNet funcionantes y no funcionantes, el otro porcentaje de causas se reduce a las extrahipofisiarias (tumores neuroendocrinos TNE, tumores hipotalámicos, hiperplasia somatotropa) y las asociadas a síndromes genéticos.
Manifestaciones clínicas
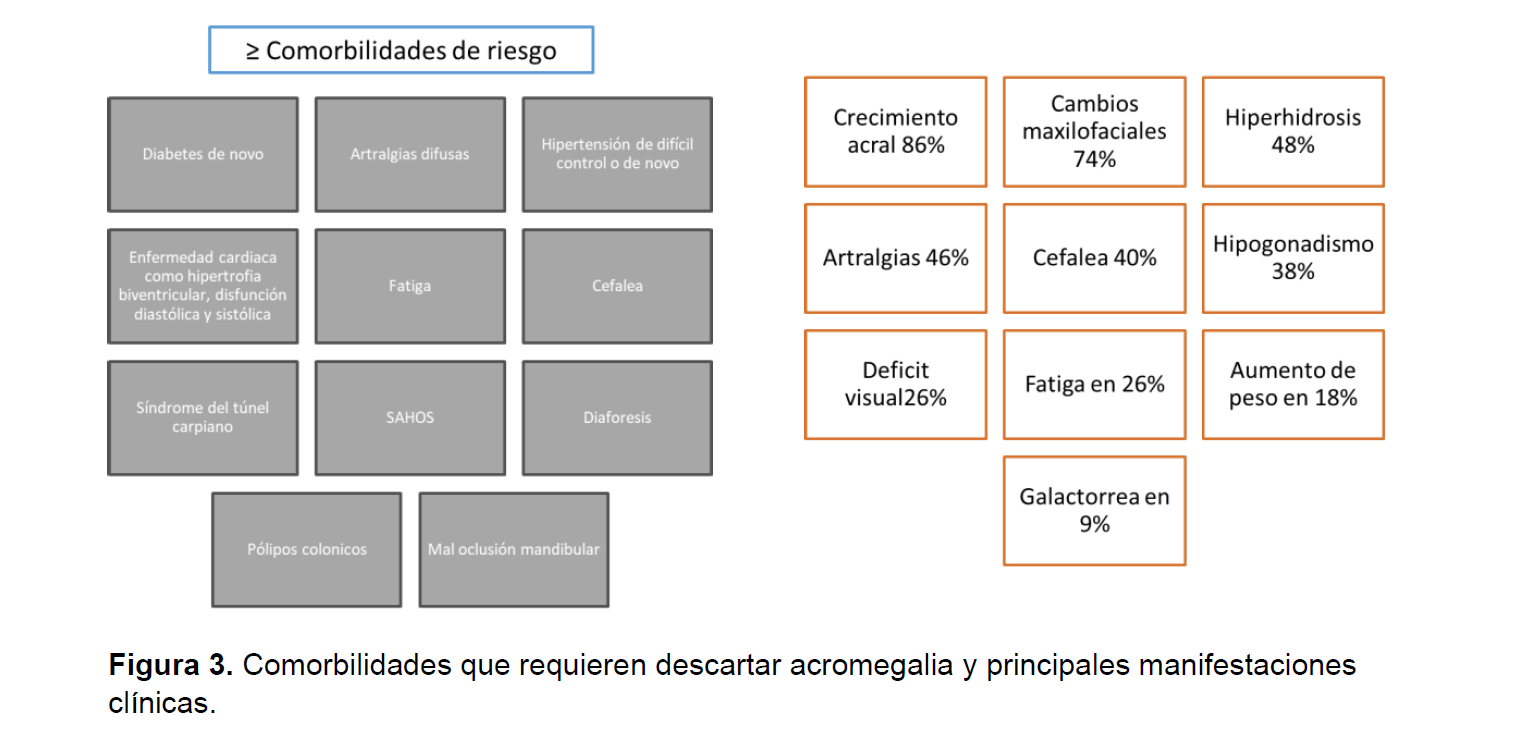
Los hallazgos al examen físico y las comorbilidades de riesgo son el primer paso para iniciar el tamizaje diagnóstico de acromegalia. El diagnostico suele tardar de 7 a 10 después de la secreción de HC y suelen estar asociados con el tamaño tumoral y una mayor actividad mitótica, es fundamental para el clínico tener claridad de cuales son las manifestaciones clínicas reversibles en los pacientes con acromegalia ( ver figuras3-4) (2, 7).
Tratamiento
Las metas al definir en manejo ya sea quirúrgico o farmacológico deben enfocarse en control bioquímico de la hormona del crecimiento (GH) y el factor de crecimiento de la insulina-1 (IGF-1) para la reducción del riesgo de mortalidad.
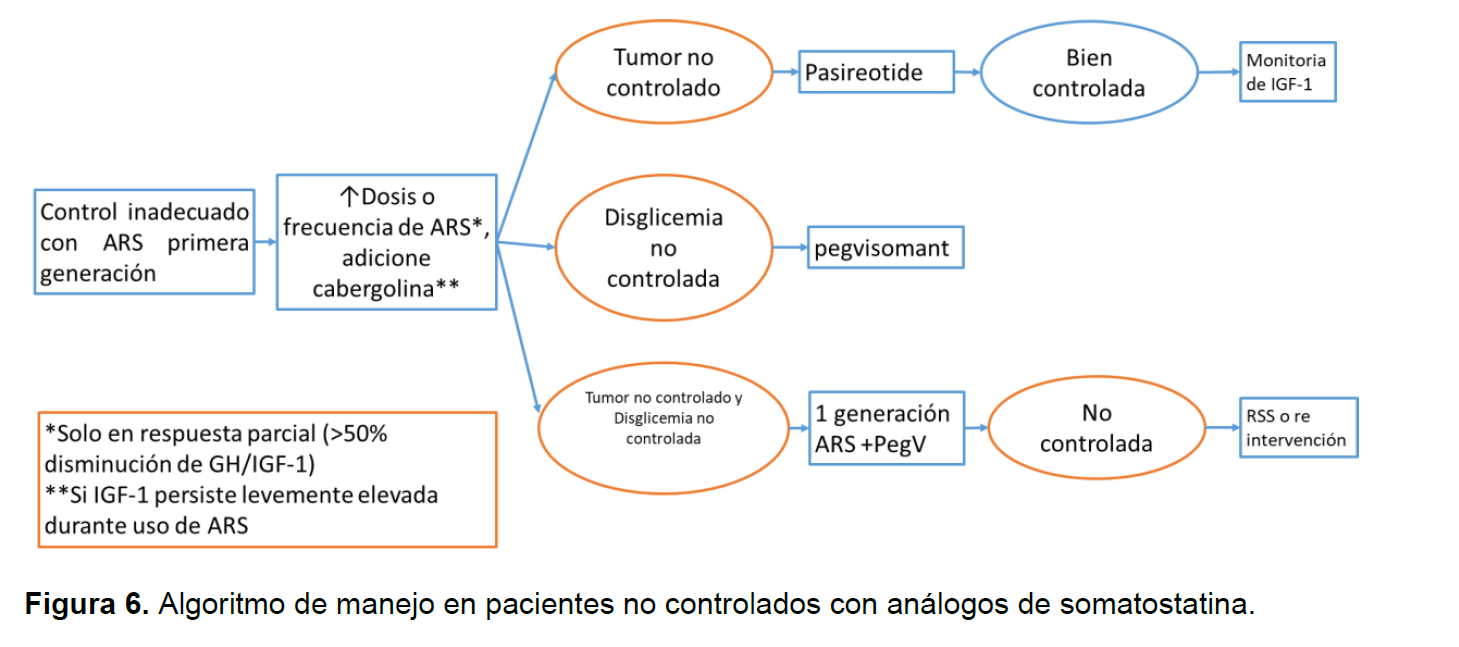
La cirugía es el tratamiento de elección para pacientes con microadenomas, macroadenomas no invasivos o macroadenomas con efecto de masa, se asociada con alta tasa de curación, baja recurrencia y pocas complicaciones, en algunos casos de manejo con citoreducción puede mejorar la respuesta hormonal a los análogos de la somatostatina (SSA), se debe pensar en manejo medico si no responde a la cirugía, si es un mal candidato quirúrgico o si es un macroadenoma sin efectos de masa y es poco probable que se cure con cirugía; la radioterapia: se usa en pacientes que no responden completamente a cirugía y/o la terapia médica (radiocirugía estereotáctica sobre la radioterapia convencional, a menos que haya una carga tumoral sustancial o un tumor <5 mm del quiasma óptico)(8).
Diagnostico
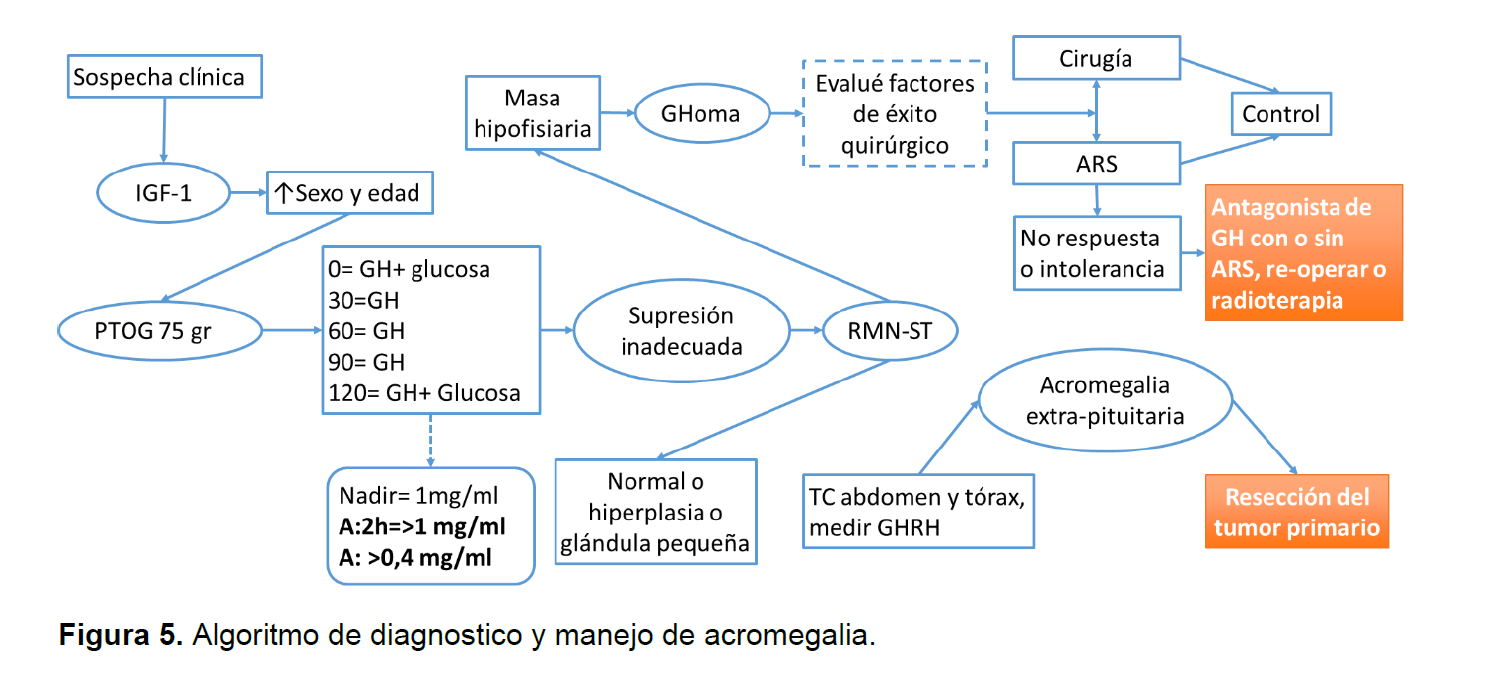
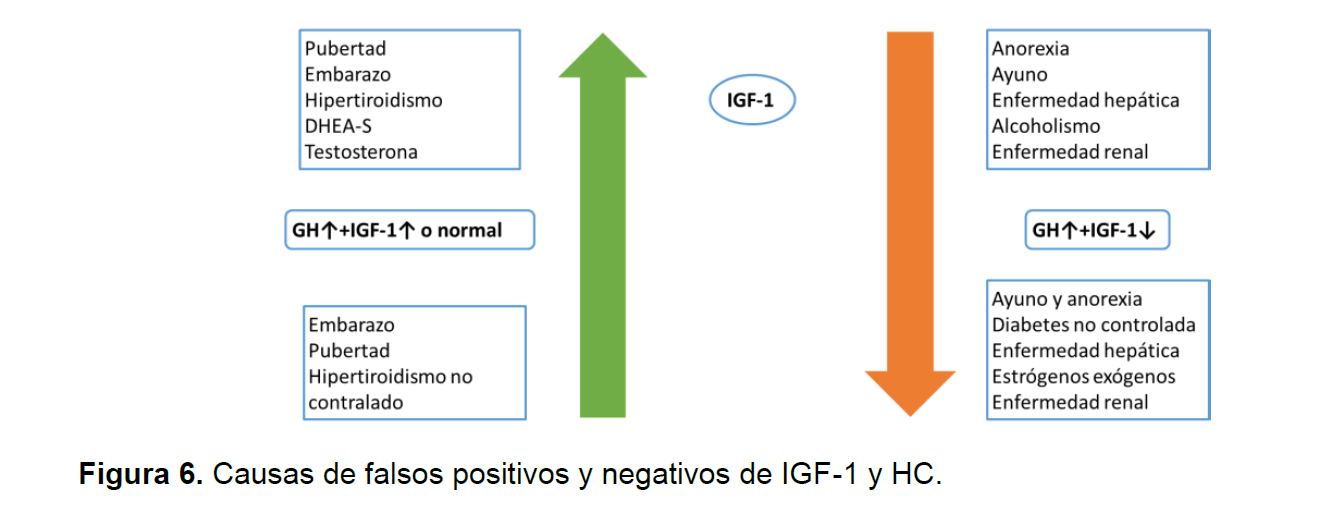
Inicialmente la prueba de tamizaje es la IGF-1 sin embargo una vez se sospeche la acroemegalia se debe hacer pruebas confirmatorias de dinámica hormonal con estimulo, es importante para el clínico recordar potenciales causas de diminución de IGF-1 y HC y aumento de IGF-1 y HC (Ver figura 5-6)(6).
Conclusiones
- Poco prevalentes, no olvidar causas no hipofisarias.
- Expresión larvada y no siempre es con síntomas floridos.
- Cirugía TE tiene buenos resultados.
- ARS son una buena opción en adyuvancia y como terapia.
- Pegvisomant y AD son excelentes coadyuvantes.
- La radioterapia es una opción con altas complicaciones.
Referencias:
- Colao A, Grasso LF, Giustina A, Melmed S, Chanson P, Pereira AM, et al. Acromegaly. Nature Reviews Disease Primers. 2019;5(1):1-17.
- Melmed S, Polonsky KS, Larsen PR, Kronenberg HM. Williams Textbook of Endocrinology E-Book: Elsevier Health Sciences; 2015.
- Ben-Shlomo A, Melmed S. Acromegaly. Endocrinology and metabolism clinics of North America. 2008;37(1):101-22.
- Rojas W. Actualización en Acromegalia. Revista Repertorio de Medicina y Cirugía. 2003;12(2):59-66.
- Yavropoulou MP, Tsoli M, Barkas K, Kaltsas G, Grossman A. The natural history and treatment of non-functioning pituitary adenomas (non-functioning PitNETs). Endocrine-Related Cancer. 2020;27(10):R375-R90.
- Melmed S. Pituitary-tumor endocrinopathies. New England Journal of Medicine. 2020;382(10):937-50.
- Carroll PV, Jenkins PJ. Acromegaly. 2015.
- Katznelson L, Atkinson JL, Cook DM, Ezzat SZ, Hamrahian AH, Miller KK, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of acromegaly-2011 update. Endocrine practice. 2011;17:1-44.
El presente material médico-científico tiene fines educativos, está dirigido única y exclusivamente a profesionales del área de la salud y está protegido por derechos de autor. Confidencial – Pfizer, todos los derechos reservados. Prohibida su reproducción total o parcial sin autorización del titular.
EM-COL-RAD-0077